Allows to interactively explore and analyze, both taxonomically and functionally, large-scale microbiome sequencing data. MEGAN is a comprehensive microbiome analysis toolbox for metagenome, metatranscriptome, amplicon and from other sources data. User can perform taxonomic, functional or comparative analysis, map reads to reference sequences, reference-based multiple alignments and reference-guided assembly and integrate their own classifications.
However, the main application of the program is to parse and analyze the result of an alignment of a set of reads against one or more reference databases, typically using BLASTN, BLASTX or BLASTP or similar tools to compare against NCBI-NT, NCBI-NR or genome specific databases. The result of a such an analysis is an estimation of the taxonomical content (“species profile”) of the sample from which the reads were collected. The program uses a number of different algorithms to “place” reads into the taxonomy by assigning each read to a taxon at some level in the NCBI hierarchy, based on their hits to known sequences, as recorded in the alignment file.
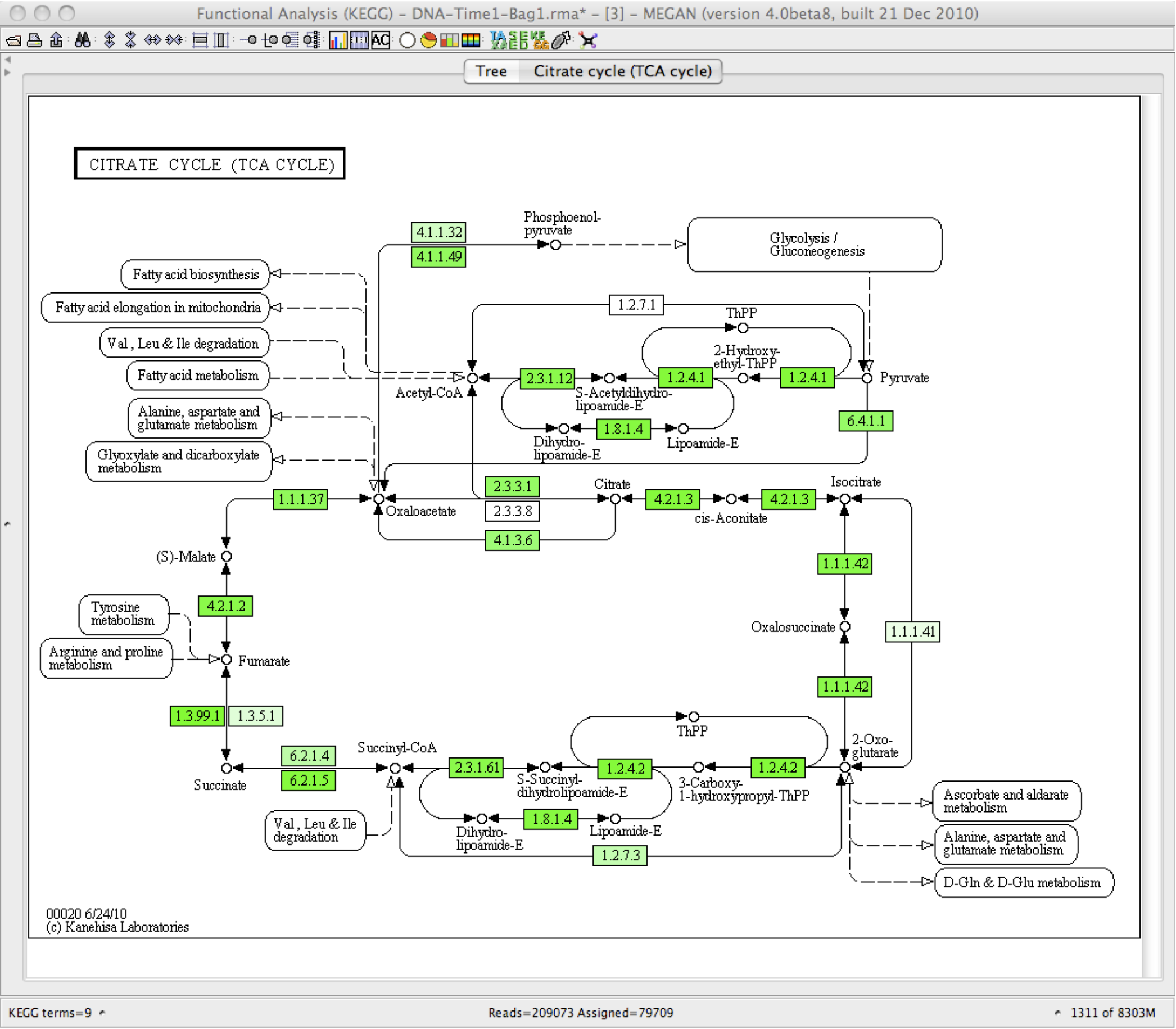
Alternatively, MEGAN can also parse files generated by the RDP website or the Silva website. Moreover, MEGAN can parse files in SAM format. MEGAN provides functional analysis using a number of different classification systems. The InterPro2GO viewer is based on the Gene Ontology metagenomic goslim and the InterPro database. The eggNOG viewer is based on the “eggNOG” database of orthologous groups and functional annotation.
Detailed information: Assigning Reads to Taxa - LCA-assignment algorithms
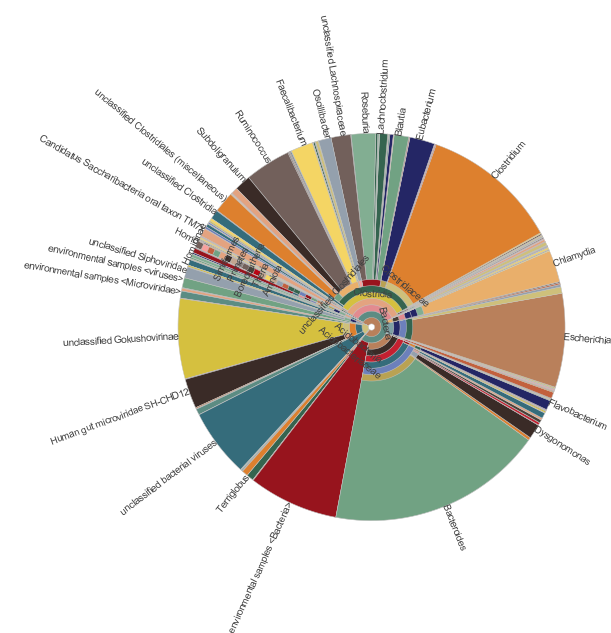
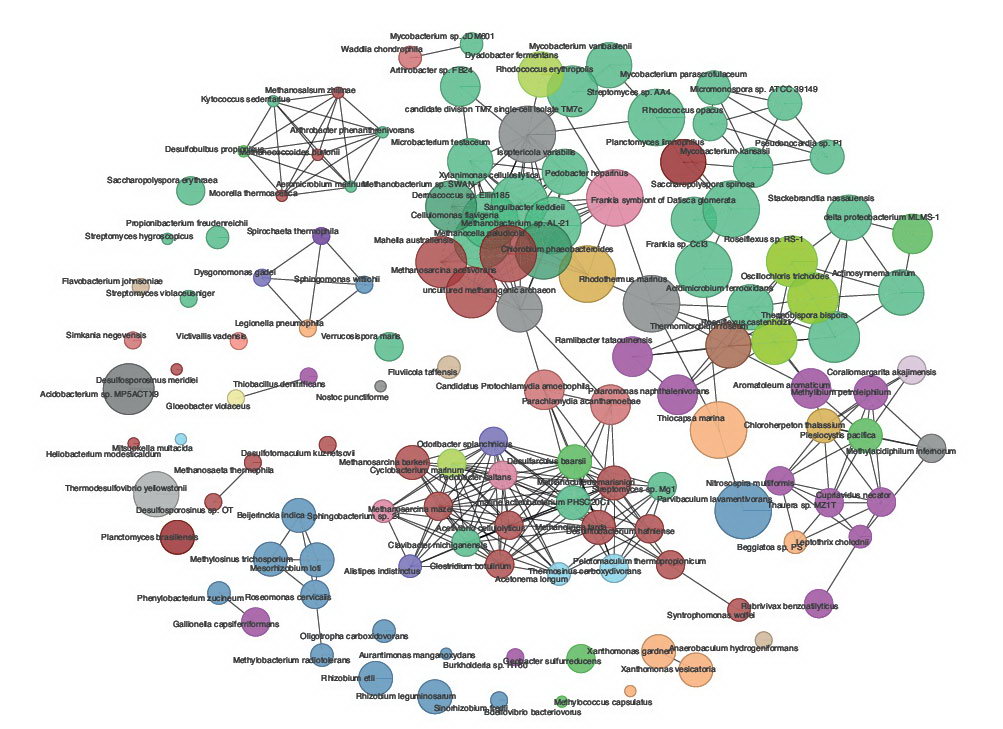
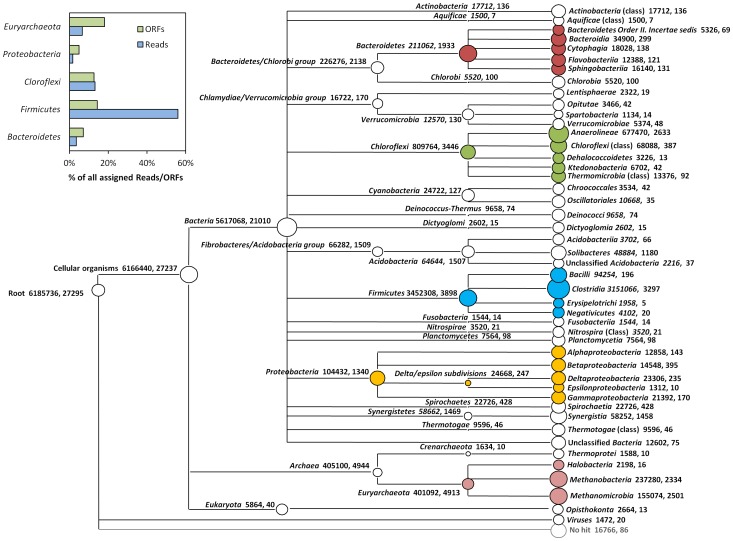
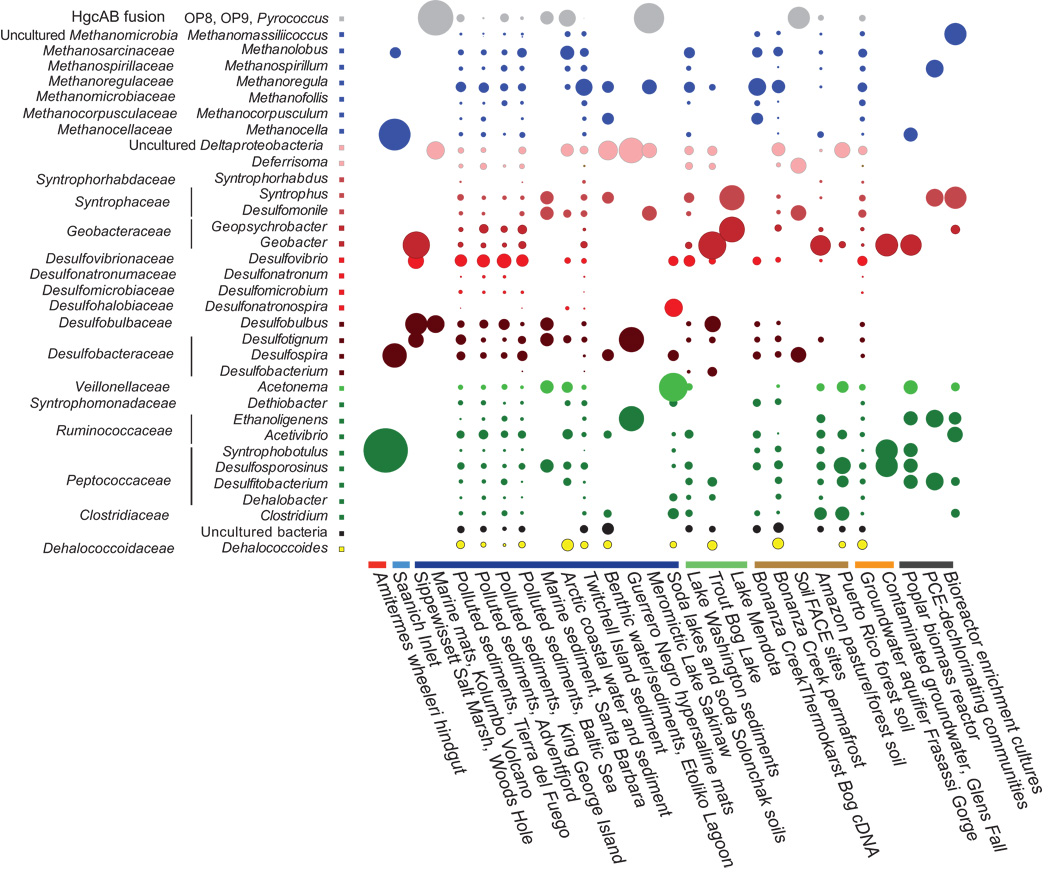
Some example of the various visualisations generated by MEGAN:
 |
 |
 |
 |
 |
 |